QC_adult
Karissa Barthelson
2024-11-01
Last updated: 2024-11-01
Checks: 7 0
Knit directory:
2021_MPSIIIBvQ96-RNAseq-7dpfLarve/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20211120) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 7bb9ce6. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rapp.history
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: code/.DS_Store
Ignored: code/fromHPC/.DS_Store
Ignored: code/fromHPC/smk/.DS_Store
Ignored: code/fromHPC/smk/slurm/.DS_Store
Ignored: data/.DS_Store
Ignored: data/Nhi_data/.DS_Store
Ignored: data/R_objects/.DS_Store
Ignored: data/R_objects/larvae/.DS_Store
Ignored: data/adult_brain/.DS_Store
Ignored: data/adult_brain/05_featureCounts/.DS_Store
Ignored: data/adult_brain/fastqc_raw/.DS_Store
Ignored: data/gene_sets/.DS_Store
Ignored: data/larvae/.DS_Store
Ignored: data/larvae/fastqc_align/.DS_Store
Ignored: data/larvae/fastqc_align_dedup/.DS_Store
Ignored: data/larvae/fastqc_raw/.DS_Store
Ignored: data/larvae/fastqc_trim/.DS_Store
Ignored: data/larvae/featureCounts/.DS_Store
Ignored: data/larvae/meta/.DS_Store
Ignored: data/larvae/starAlignLog/.DS_Store
Ignored: output/.DS_Store
Ignored: output/plots/
Ignored: output/plots4poster/.DS_Store
Ignored: output/plots4pub/
Untracked files:
Untracked: Rplot01.pdf
Untracked: Rplot02.png
Untracked: code/6mBrain_RNAseq_genoSex.rmd
Untracked: code/6mBrain_fems.rmd
Untracked: code/HS.R
Untracked: code/about.Rmd
Untracked: code/analysis.Rmd
Untracked: code/analysis_final.rmd
Untracked: code/analysis_final_v2.rmd
Untracked: code/checkGenotypes.rmd
Untracked: code/explorationRUV.Rmd
Untracked: code/genotypeCheck.Rmd
Untracked: code/license.Rmd
Untracked: code/nhi6mdata.rmd
Untracked: code/plots4pub2.rmd
Untracked: code/plots4pub_RNAseq_afterreview1.R
Untracked: code/wgcna.rmd
Untracked: data/R_objects/adult_brain/HMPKEGG_6m_brain_genosex_CQN.rds
Untracked: data/R_objects/adult_brain/HMP_ire_6m_brain_genosex_CQN.rds
Untracked: data/R_objects/adult_brain/cqn_logCPM_6m_brain.rds
Untracked: data/R_objects/adult_brain/dge_6m_brain_cqn.rds
Untracked: data/R_objects/adult_brain/fry_cell_6m_brain_genosex_CQN.rds
Untracked: data/R_objects/adult_brain/fry_cell_RNAseq.rds
Untracked: data/R_objects/adult_brain/goseqtibbles_6m_brain_genosex_CQN.rds
Untracked: data/R_objects/adult_brain/hmp.kegg.rna.rds
Untracked: data/R_objects/adult_brain/toptablescqn_bysex.rds
Untracked: data/allLLIDs.rds
Untracked: data/ensembl_zebrafish.rds
Untracked: data/proteomics/
Untracked: data/with_go_evidence.rds
Untracked: dre00020.pathview.multi.png
Untracked: dre00020.png
Untracked: dre00020.xml
Untracked: hsa04650.EOfADproteomes.multi.png
Untracked: hsa04650.MPSIIIBproteomes.multi.png
Untracked: hsa04650.png
Untracked: hsa04650.xml
Untracked: hsa04666.EOfADproteomes.multi.png
Untracked: hsa04666.MPSIIIBproteomes.multi.png
Untracked: hsa04666.png
Untracked: hsa04666.xml
Untracked: larvae dre00020.pathview.multi.png
Untracked: output/DE_proteins_for_string/
Untracked: output/GOenrichment_enrichmentTable.csv
Untracked: output/plots4RADposter/
Untracked: output/plots4kim/
Untracked: output/plots4pub2/
Untracked: output/plots4pub3/
Untracked: output/spreadsheets/RNAseqlarvae_celltypeFry.xlsx
Untracked: output/spreadsheets/RNAseqlarvae_hmpoutputs.xlsx
Untracked: output/spreadsheets/geneset_RNAseq_v2.xlsx
Untracked: output/spreadsheets/goseq_RNAseq_v2.xlsx
Untracked: output/spreadsheets/limma_proteomics_6mbrain.xlsx
Untracked: output/spreadsheets/proteomics_6mbrain_hmpoutputs.xlsx
Untracked: output/spreadsheets/toptables_RNAseq_v2.xlsx
Untracked: temp.png
Unstaged changes:
Deleted: analysis/6mBrain_fems.rmd
Deleted: analysis/HS.R
Deleted: analysis/about.Rmd
Deleted: analysis/analysis.Rmd
Deleted: analysis/analysis_adultbrain.knit.md
Deleted: analysis/analysis_final.rmd
Deleted: analysis/checkGenotypes.rmd
Deleted: analysis/explorationRUV.Rmd
Deleted: analysis/genotypeCheck.Rmd
Deleted: analysis/license.Rmd
Deleted: analysis/nhi6mdata.rmd
Deleted: analysis/plots4pub2.rmd
Modified: data/R_objects/adult_brain/dge.rds
Modified: data/R_objects/adult_brain/hmp_ire.rds
Modified: data/R_objects/adult_brain/logcpm.rds
Modified: data/R_objects/larvae/celltype_larvae.rds
Modified: data/R_objects/larvae/dge.rds
Modified: data/R_objects/larvae/hmp_ire.rds
Modified: data/R_objects/larvae/hmp_kegg.rds
Modified: data/R_objects/larvae/logcpm.rds
Modified: data/R_objects/larvae/toptablescqn.rds
Modified: data/adult_brain/karissas_metadata.xlsx
Modified: output/spreadsheets/toptables_cqn.xlsx
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/QC_adult.rmd) and HTML
(docs/QC_adult.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 7bb9ce6 | Karissa Barthelson | 2024-11-01 | wflow_publish("analysis/*") |
Introduction
library(tidyverse)
library(magrittr)
library(readxl)
library(ngsReports)
library(plotly)
library(AnnotationHub)
library(pander)
library(scales)
library(pheatmap)
library(ggpubr)
theme_set(theme_bw())ah <- AnnotationHub() %>%
subset(species == "Danio rerio") %>%
subset(rdataclass == "EnsDb")
ensDb <- ah[["AH83189"]] # for release 101, latest version and the alignment
grTrans <- transcripts(ensDb)
trLengths <- exonsBy(ensDb, "tx") %>%
width() %>%
vapply(sum, integer(1))
mcols(grTrans)$length <- trLengths[names(grTrans)]
gcGene <- grTrans %>%
mcols() %>%
as.data.frame() %>%
dplyr::select(gene_id, tx_id, gc_content, length) %>%
as_tibble() %>%
group_by(gene_id) %>%
summarise(
gc_content = sum(gc_content*length) / sum(length),
length = ceiling(median(length))
)
grGenes <- genes(ensDb)
mcols(grGenes) %<>%
as.data.frame() %>%
left_join(gcGene) %>%
as.data.frame() %>%
DataFrame()# readin meta and tidy up columns
meta <- read_excel("data/adult_brain/karissas_metadata.xlsx", sheet = "onlyseq") %>%
mutate(genotype = case_when(
`usable genotype?` == "wt" ~ "wt",
`usable genotype?` == "EOfAD" ~ "EOfADlike",
`usable genotype?` == "MPS-III" ~ "MPSIIIB"
) %>%
factor(levels = c("wt", "EOfADlike", "MPSIIIB")),
tank = as.factor(tank),
sex = as.factor(sex),
sample = paste0(fish, "_", genotype),
group = paste0(genotype, "_", sex) %>% as.factor(),
age = "6 m adult brain",
RIN = as.numeric(`RIN/DIN`)
) %>%
dplyr::select(
# reorder the metadata nicely
fish,genotype,sex,group,batch,tank,RIN,everything()
) %>%
as_tibble()Here, I will assess the quality of the RNA-seq data for the psen1 Q96_K97del/+ vs naglu A603fs/A603fs experiment on zebrafish brains at 6 mo old.
Total RNA was purified from the brains of individual fish, while the tail end was used for gDNA extraction and PCR genotyping. The total RNA was DNase treated (to remove any genomic DNA which was carried over from the RNA extraction), then delivered to SAGC for polyA+ library preparation and sequencing using the MGI DNBSEQ technology.
The sequencing was performed over four lanes which were subsequently
merged. This was done using the merge.sh script shown
below.
## Insert the merge files script here
readLines("code/mergeFiles.sh") %>%
cat(sep = "\n")#!/bin/bash
#SBATCH -p batch
#SBATCH -N 1
#SBATCH -n 8
#SBATCH --time=0-01:00:00
#SBATCH --mem=16GB
#SBATCH --mail-type=END
#SBATCH --mail-type=FAIL
#SBATCH --mail-user=karissa.barthelson@adelaide.edu.au
##Params
mkdir /hpcfs/users/a1211024/q96-v-naglu7dpf/temp
FASTDATA1=/hpcfs/users/a1211024/q96-v-naglu7dpf/V350030606
TEMP=/hpcfs/users/a1211024/q96-v-naglu7dpf/temp
FASTOUT=/hpcfs/users/a1211024/q96-v-naglu7dpf/fastq
## Concatenating the F reads
for R1 in ${FASTDATA1}/*_1_R1_001.fastq.gz
do
# Define the other lanes
R2=${R1%_1_R1_001.fastq.gz}_2_R1_001.fastq.gz
R3=${R1%_1_R1_001.fastq.gz}_3_R1_001.fastq.gz
R4=${R1%_1_R1_001.fastq.gz}_4_R1_001.fastq.gz
CATNAME=$(basename ${R1%_1_R1_001.fastq.gz})
echo -e "cat will merge:\t${R1}\n\t${R2}\n\t${R3}\n\t${R4}"
echo -e "New file name will be:\t${TEMP}/${CATNAME}_merged_R1_001.fastq.gz"
cat ${R1} ${R2} ${R3} ${R4} > ${TEMP}/${CATNAME}_merged_R1_001.fastq.gz
done
## Concatenating the R reads
for R1 in ${FASTDATA1}/*_1_R2_001.fastq.gz
do
# Define the other lanes
R2=${R1%_1_R2_001.fastq.gz}_2_R2_001.fastq.gz
R3=${R1%_1_R2_001.fastq.gz}_3_R2_001.fastq.gz
R4=${R1%_1_R2_001.fastq.gz}_4_R2_001.fastq.gz
CATNAME=$(basename ${R1%_1_R2_001.fastq.gz})
echo -e "cat will merge:\t${R1}\n\t${R2}\n\t${R3}\n\t${R4}"
echo -e "New file name will be:\t${TEMP}/${CATNAME}_merged_R2_001.fastq.gz"
cat ${R1} ${R2} ${R3} ${R4} > ${TEMP}/${CATNAME}_merged_R2_001.fastq.gz
done
# ## Move the merged files from temp - fastq
mkdir 01_rawdata
mkdir 01_rawdata/fastq
mv ${TEMP}/*fastq.gz 01_rawdata/fastq
# remove the temp files/dirs
rm ${TEMP}/*.*
rmdir ${TEMP}fastqc: raw data
Here, I will use the ngsReports package to combine and
visualise the fastqc results.
fastqc_raw <- list.files(
path = "data/adult_brain/fastqc_raw",
pattern = "zip",
recursive = TRUE,
full.names = TRUE) %>%
FastqcDataList()The total number of reads ranged between 24,185,951 and
78,793,504 reads. Note that the number of reads in the
R1 file indeed equals to the number of reads in the
R2 file.
readTotals(fastqc_raw) %>%
mutate(Read = case_when(
grepl(Filename, pattern = "_R1") ~ "R1",
grepl(Filename, pattern = "_R2") ~ "R2"
),
ULN = str_remove(Filename, "_S.+_merged.+")
) %>%
left_join(meta) %>%
ggplot(aes(x = ULN, y = Total_Sequences, fill = Read)) +
geom_col(position = "dodge") +
coord_flip() +
scale_fill_viridis_d(end = 0.8) +
facet_wrap(~genotype, scales = "free_y", ncol = 1, strip.position = "right")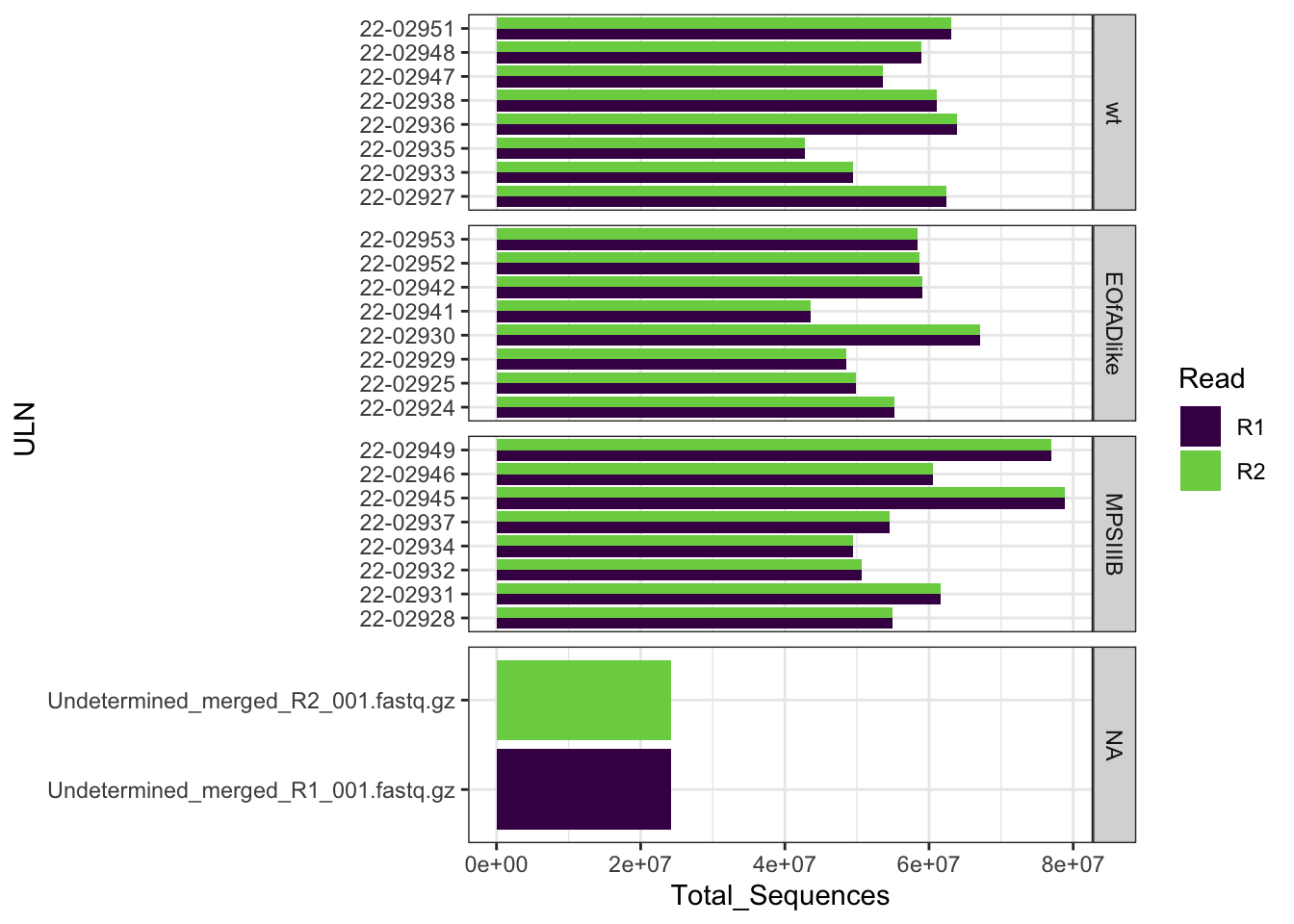
The base quality of all the reads also looked good.
plotBaseQuals(fastqc_raw)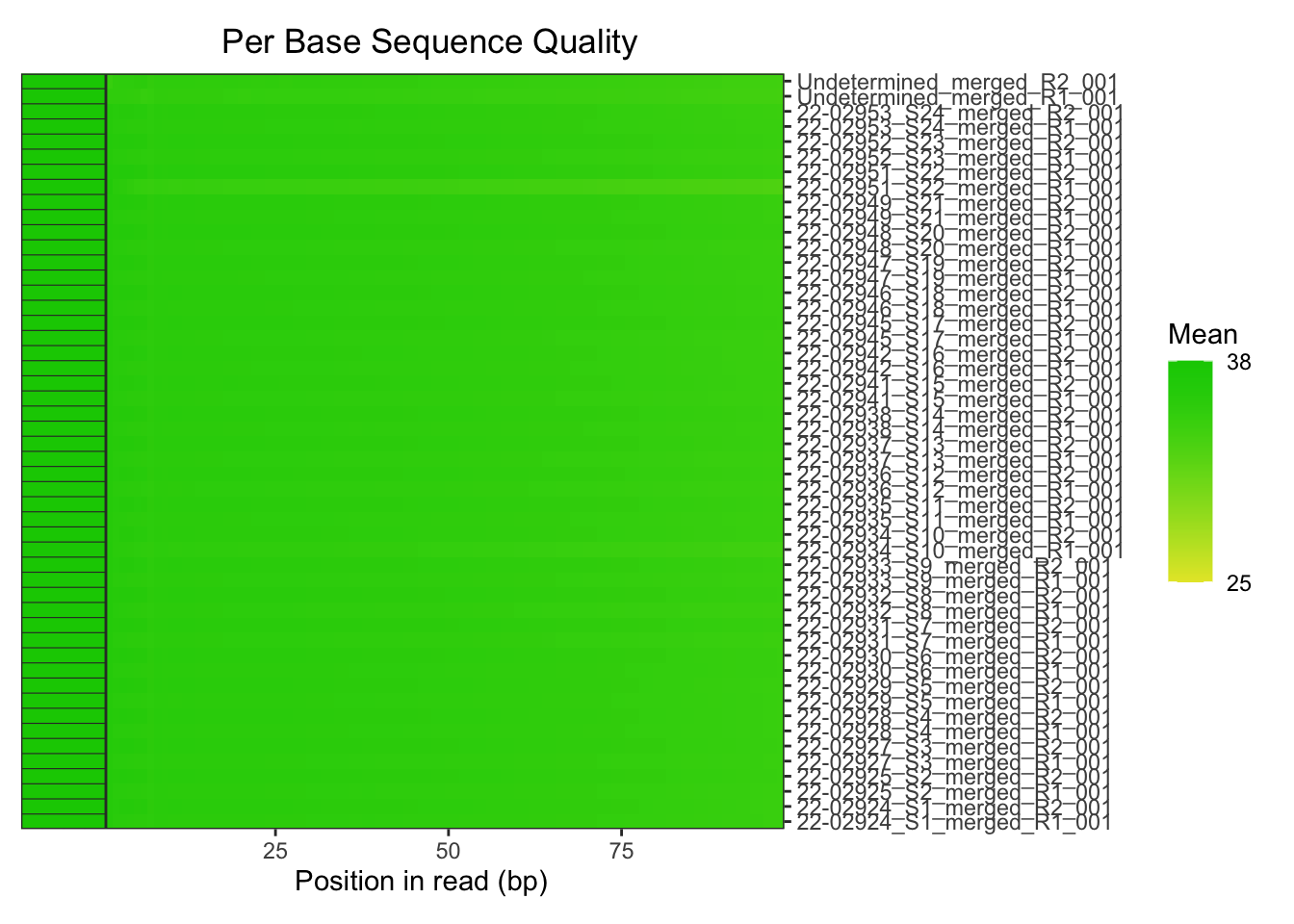
GC Content
All samples have similar GC content. No issues are present.
plotGcContent(
x = fastqc_raw,
plotType = "line",
gcType = "Transcriptome",
species = "Drerio",
usePlotly = TRUE
)Over-repreented seq
No over-represented sequences are present in this dataset.
getModule(fastqc_raw, "Overrep") # A tibble: 0 × 0trimmed data fastQC
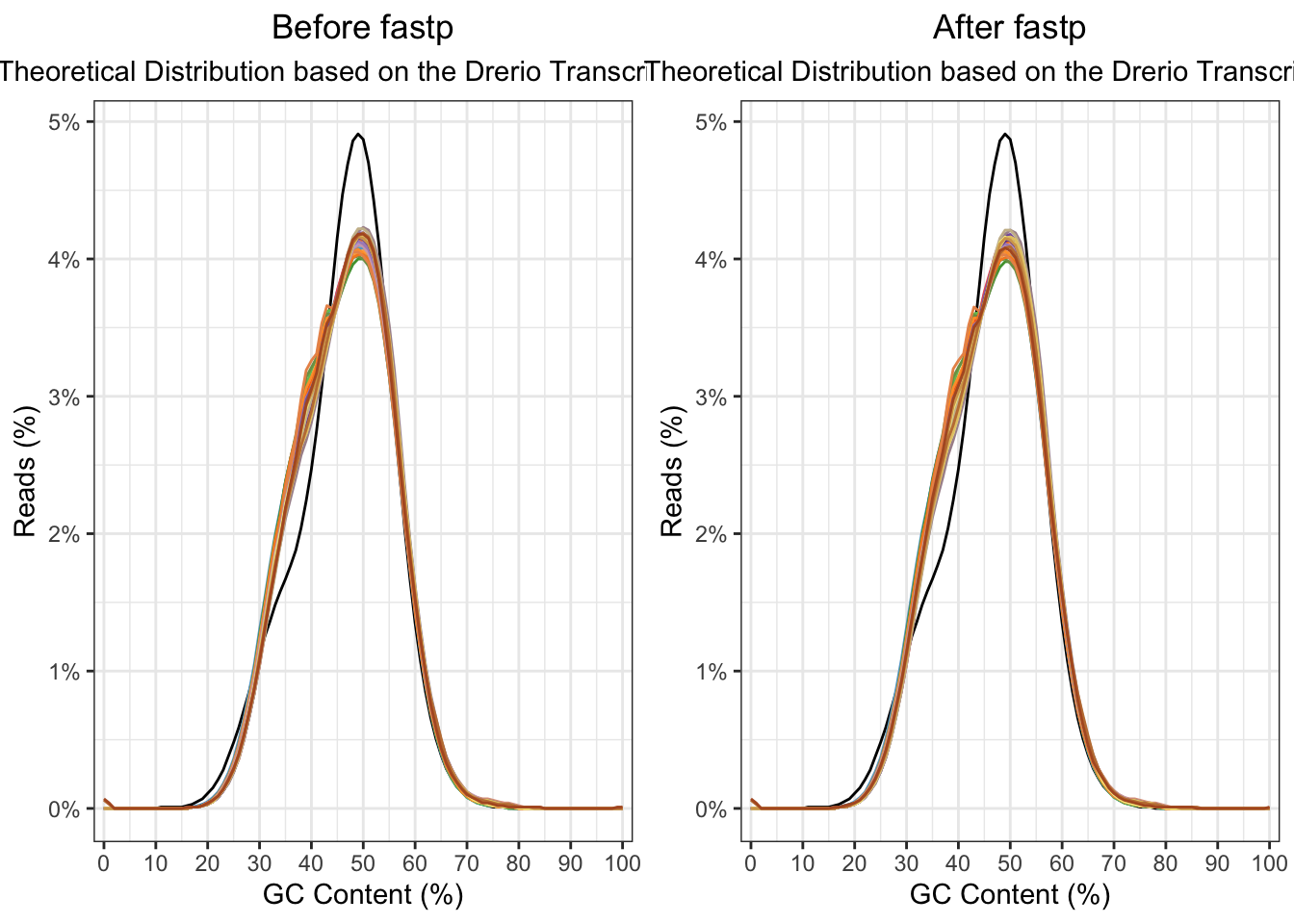
The raw fastq. files were then processed with fastp. In
this step, the adaptor sequeces were trimmed from the reads. Then all
length and quality filters were left as default values. Less than 1% of
the reads was discarded, and no observed changes are apparent in the %GC
in the reads, and quality all looks good.
fastqc_trim <- list.files(path = "data/adult_brain/fastqc_trim",
pattern = "zip",
recursive = TRUE,
full.names = TRUE) %>%
FastqcDataList()trimStats <- readTotals(fastqc_raw) %>%
dplyr::rename(Raw = Total_Sequences) %>%
left_join(readTotals(fastqc_trim), by = "Filename") %>%
dplyr::rename(Trimmed = Total_Sequences) %>%
mutate(
Discarded = 1 - Trimmed / Raw,
Retained = Trimmed / Raw
)
trimStats %>%
mutate(ULN = str_remove(Filename, "_S.+_merged.+")
) %>%
left_join(meta) %>%
unique() %>%
ggplot(aes(y = ULN)) +
geom_col(aes(x = Discarded*100)) +
facet_wrap(~genotype, scales = "free_y", ncol = 1, strip.position = "right") +
labs(x = "Percentage reads discarded by fastp")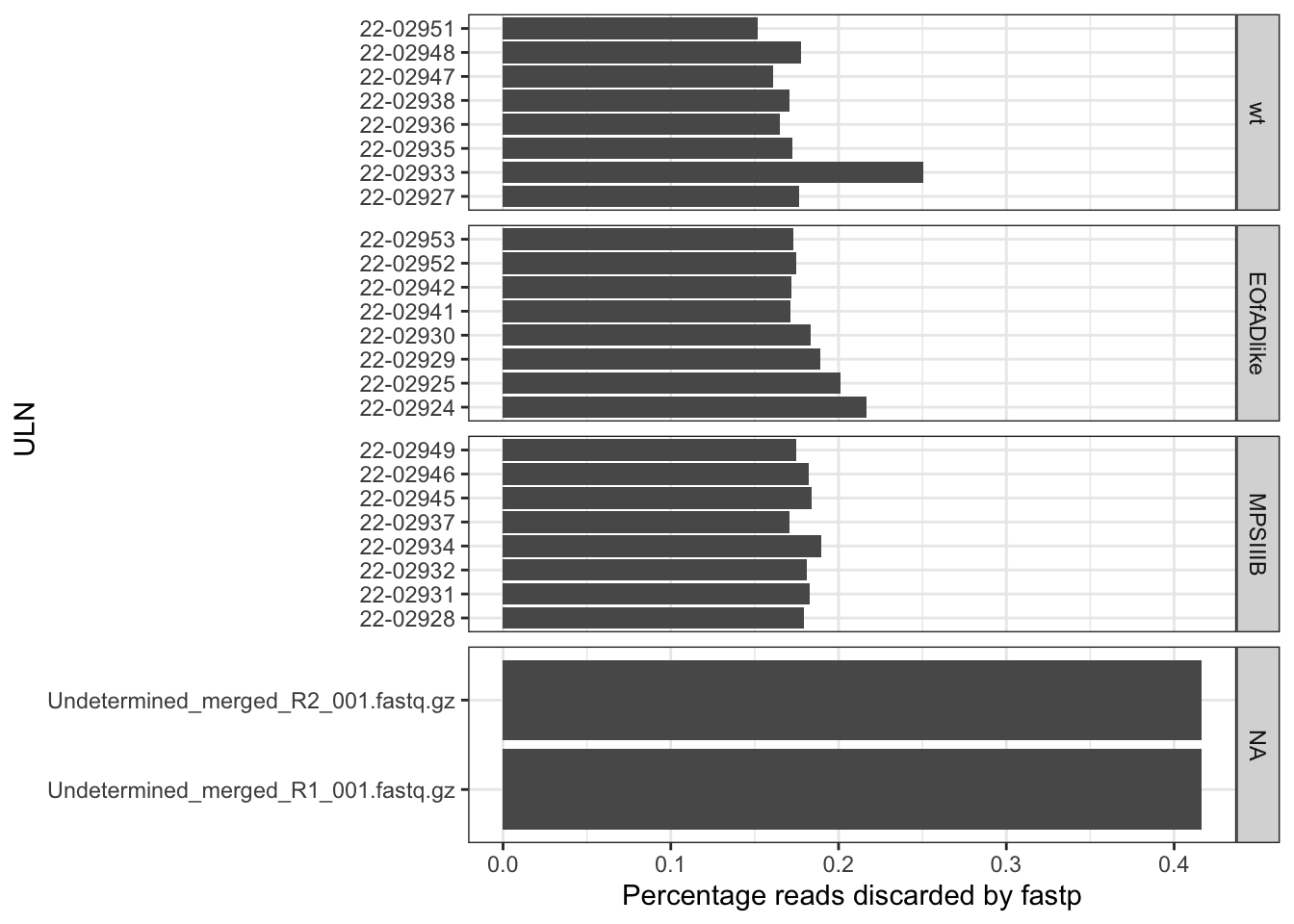
plotBaseQuals(fastqc_trim)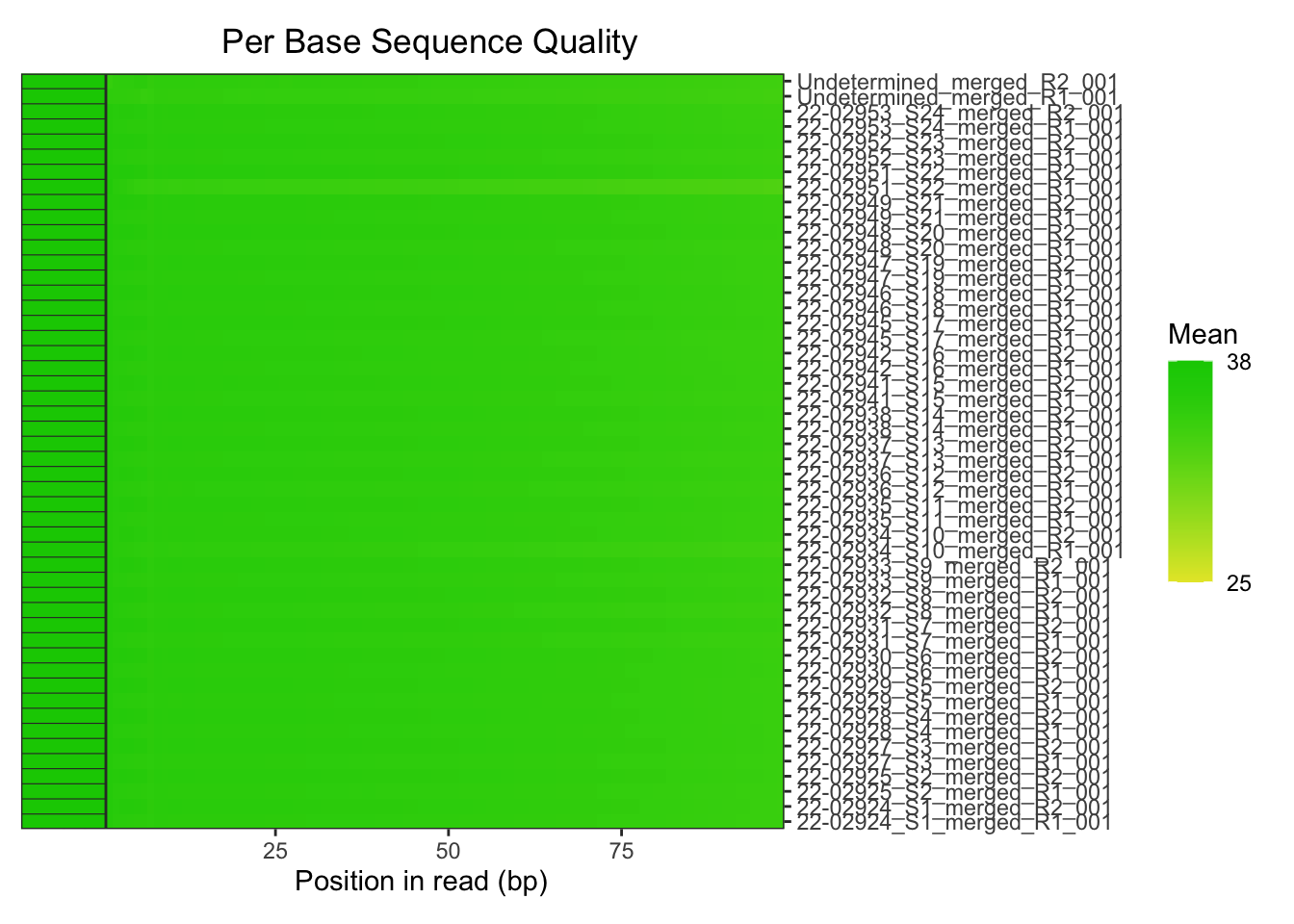
ggarrange(
plotGcContent(
x = fastqc_raw,
plotType = "line",
gcType = "Transcriptome",
species = "Drerio"
) +
theme(legend.position = "none") +
ggtitle("Before fastp"),
plotGcContent(
x = fastqc_trim,
plotType = "line",
gcType = "Transcriptome",
species = "Drerio"
) +
theme(legend.position = "none")+
ggtitle("After fastp")
) 
Aligned QC
The reads were aligned to the GRCz11 genome. The majority of reads were aligned uniquely.
fastqc_align <- list.files(
path = "data/adult_brain/fastqc_align",
pattern = "zip",
recursive = TRUE,
full.names = TRUE) %>%
FastqcDataList()list.files("data/adult_brain/starlog", full.names = TRUE) %>%
.[grepl(x = ., pattern = "Log.final.out")] %>%
ngsReports::plotAlignmentSummary(type = "star") +
scale_fill_viridis_d(end = 0.8) +
theme(legend.position = "right") +
ggtitle("Summary of alignment (STAR)",
subtitle = "In all samples, the majority of reads mapped uniquely to the zebrafish genome.")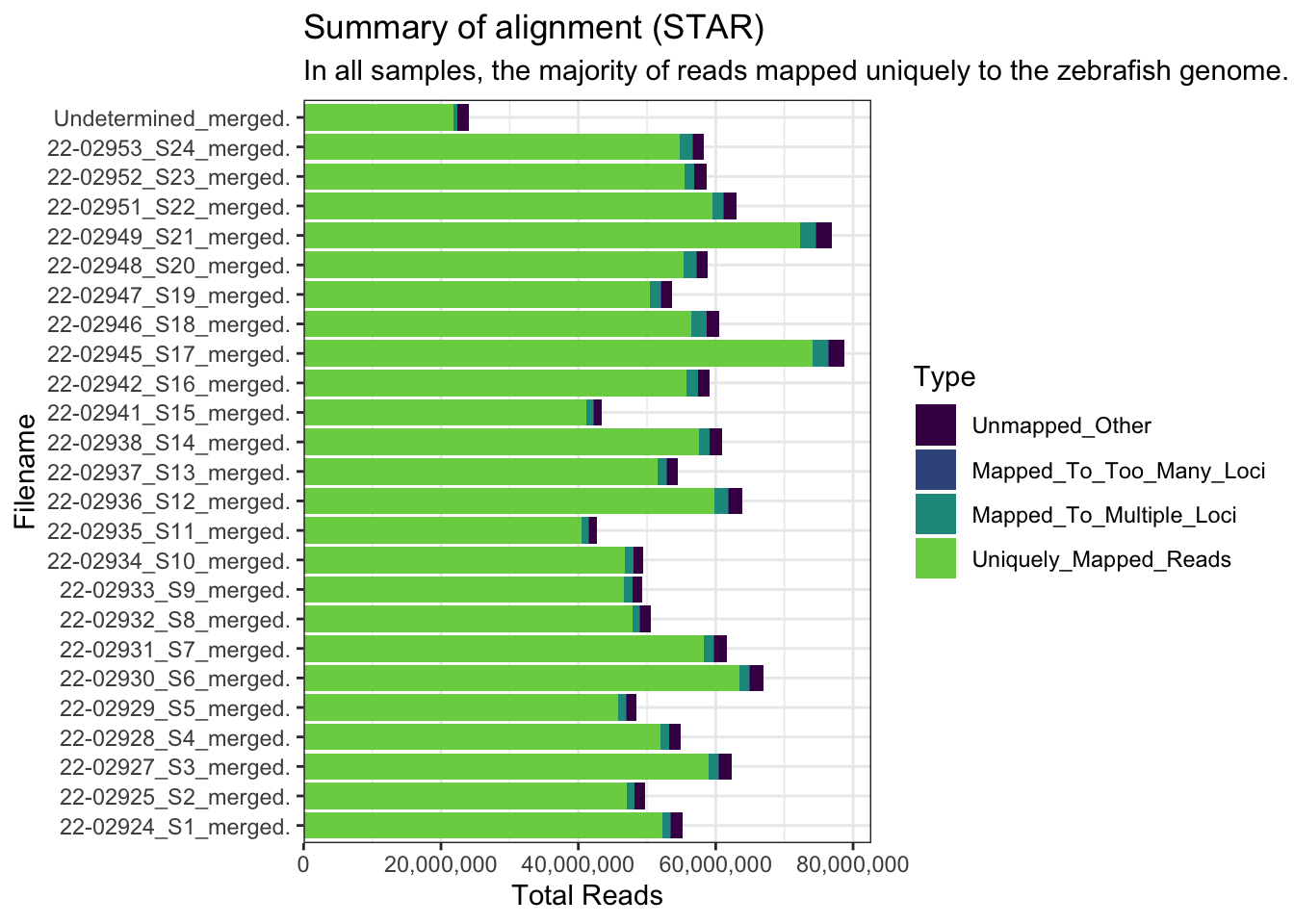
plotBaseQuals(fastqc_align)
plotGcContent(x = fastqc_align,
plotType = "line",
gcType = "Transcriptome",
species = "Drerio"
) +
theme(legend.position = "none") 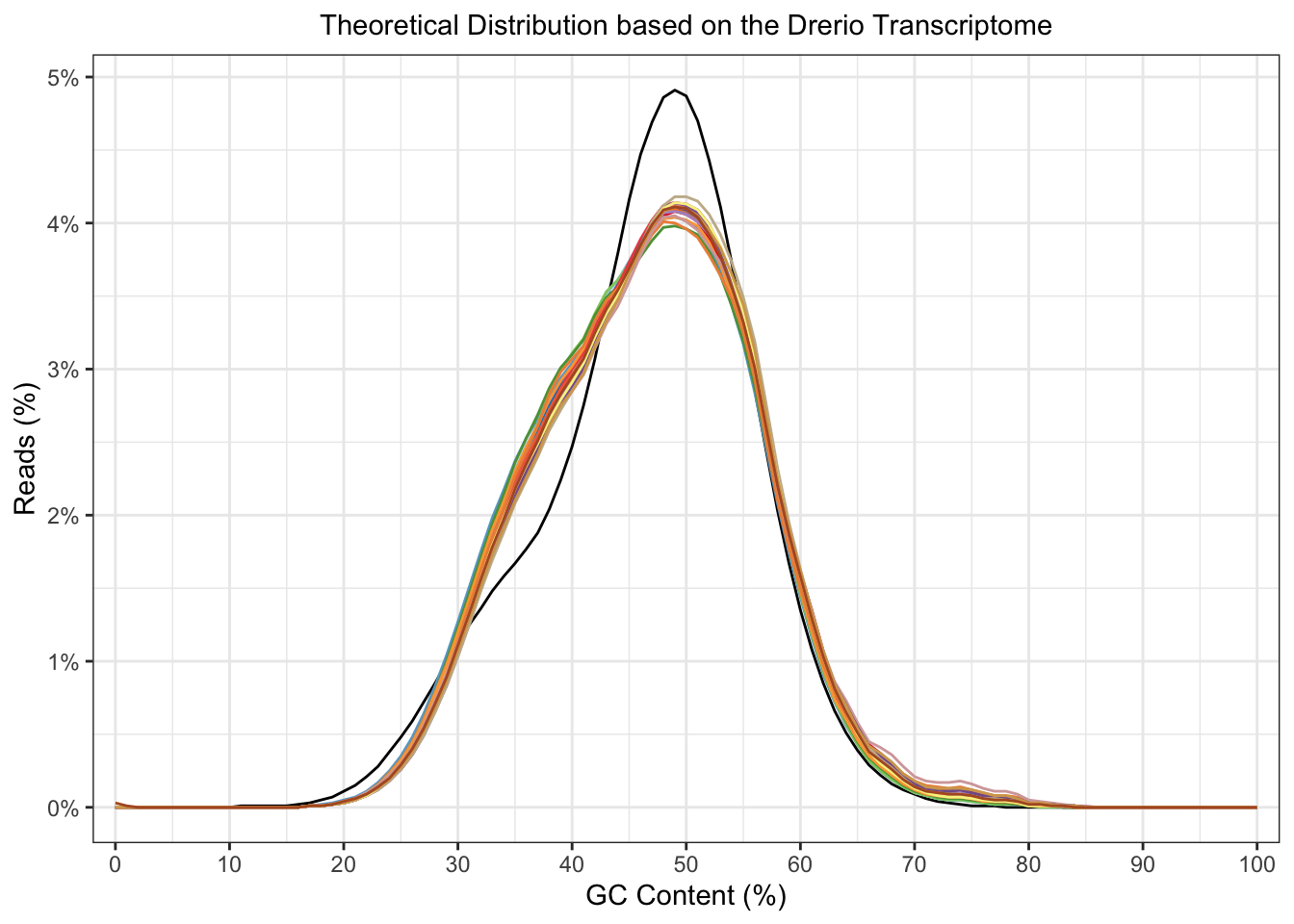
Dedup align QC
This dataset was processed with UMIs, which allow PCR duplicates to
be removed. I did this using umi-tools. After
de-duplciation ** reads were retained.
fastqc_align_dedup <- list.files(
path = "data/adult_brain/fastqc_dedup",
pattern = "zip",
recursive = TRUE,
full.names = TRUE) %>%
FastqcDataList()readTotals(fastqc_align) %>%
mutate(align = "raw") %>%
bind_rows(readTotals(fastqc_align_dedup) %>%
mutate(align = "dedup")) %>%
mutate(ULN = str_remove(Filename, "_S.+_merged.+")) %>%
left_join(meta) %>%
ggplot(aes(x = ULN, y = Total_Sequences, fill = align)) +
geom_col(position = "dodge") +
coord_flip() +
scale_fill_viridis_d(end = 0.8) +
scale_y_continuous(labels = comma) +
facet_wrap(~genotype, scales = "free_y", ncol = 1, strip.position = "right")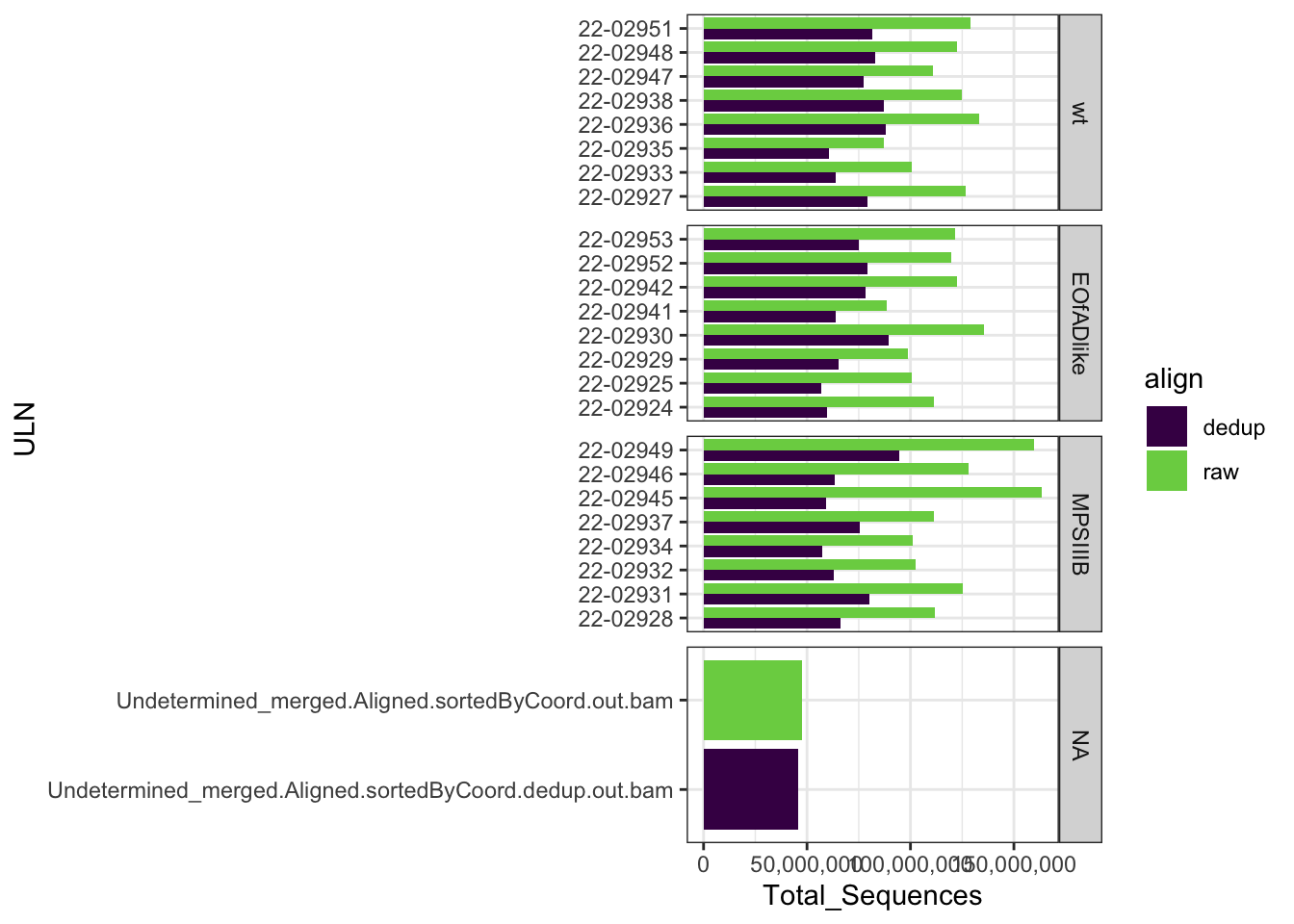
FeatureCounts summary
The majority of reads are counted unqiely.
FC_summary <-
read.delim("data/adult_brain/05_featureCounts/counts.out.summary") %>%
set_colnames(colnames(.) %>%
str_remove(pattern = "_S.+_merged.Aligned.sortedByCoord.dedup.out.bam") %>%
str_remove(pattern = "X04_dedup.bam.") %>%
str_replace(pattern = "\\.", replacement = "\\-")
)
FC_summary %>%
gather(key = "ULN", value = "NumReads", starts_with("22")) %>%
left_join(meta) %>%
as_tibble() %>%
dplyr::filter(NumReads > 0) %>%
ggplot(aes(y = ULN, x = NumReads, fill = Status)) +
geom_col() +
scale_fill_viridis_d(end = 0.8) +
scale_x_continuous(labels = comma) +
facet_wrap(~genotype, scales = "free_y", ncol = 1, strip.position = "right")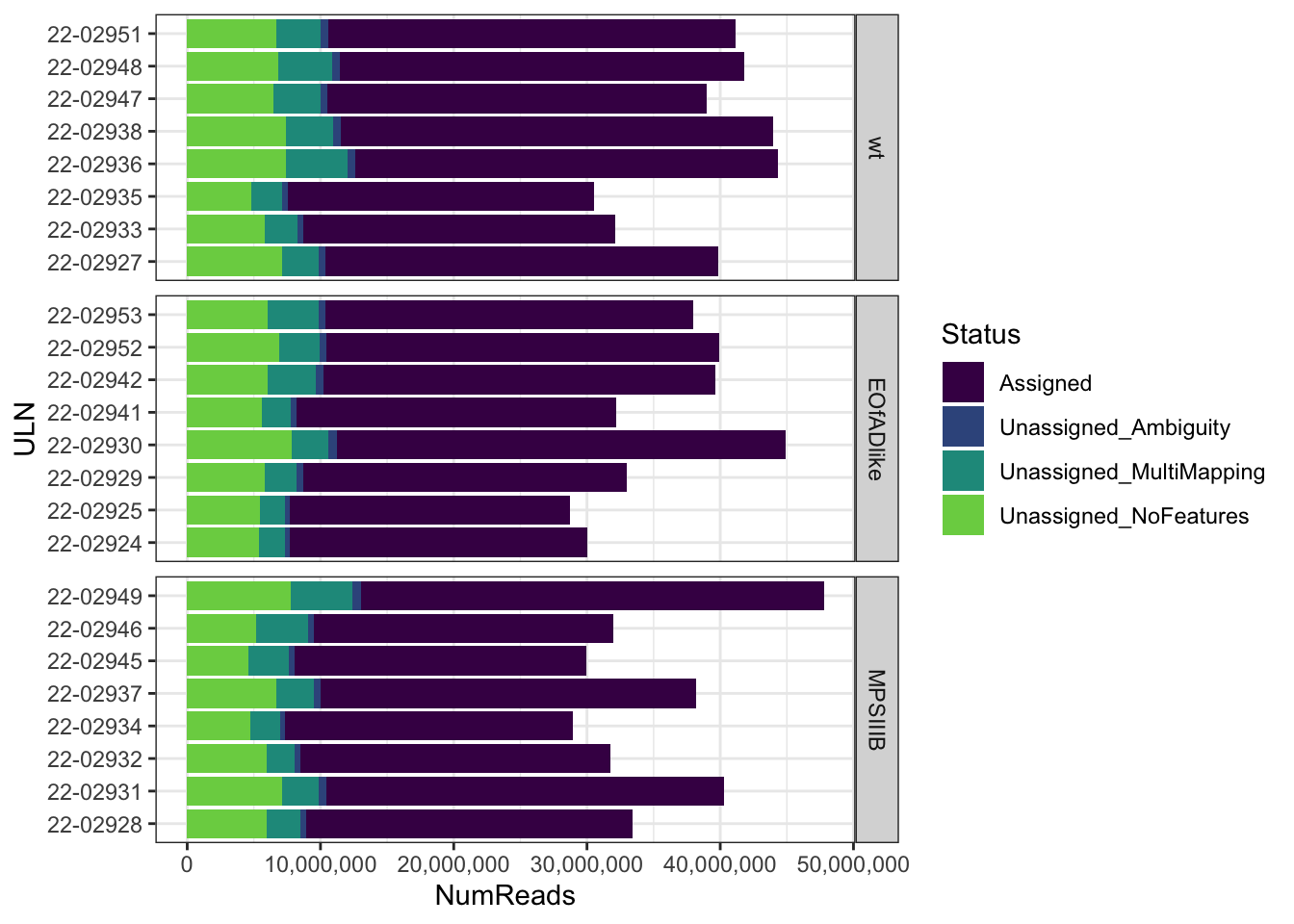
Conclusion
Data looks of sufficient quality. Proceed to analysis.
sessionInfo()R version 4.3.2 (2023-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.3
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Australia/Adelaide
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] ensembldb_2.26.0 AnnotationFilter_1.26.0 GenomicFeatures_1.54.4
[4] AnnotationDbi_1.64.1 Biobase_2.62.0 GenomicRanges_1.54.1
[7] GenomeInfoDb_1.38.8 IRanges_2.36.0 S4Vectors_0.40.2
[10] ggpubr_0.6.0 pheatmap_1.0.12 scales_1.3.0
[13] pander_0.6.5 AnnotationHub_3.10.1 BiocFileCache_2.10.2
[16] dbplyr_2.5.0 plotly_4.10.4 ngsReports_2.4.0
[19] patchwork_1.2.0 BiocGenerics_0.48.1 readxl_1.4.3
[22] magrittr_2.0.3 lubridate_1.9.3 forcats_1.0.0
[25] stringr_1.5.1 dplyr_1.1.4 purrr_1.0.2
[28] readr_2.1.5 tidyr_1.3.1 tibble_3.2.1
[31] ggplot2_3.5.0 tidyverse_2.0.0 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3 ggdendro_0.2.0
[3] rstudioapi_0.16.0 jsonlite_1.8.8
[5] farver_2.1.1 rmarkdown_2.26
[7] BiocIO_1.12.0 fs_1.6.3
[9] zlibbioc_1.48.2 vctrs_0.6.5
[11] Rsamtools_2.18.0 memoise_2.0.1
[13] RCurl_1.98-1.14 rstatix_0.7.2
[15] S4Arrays_1.2.1 htmltools_0.5.8.1
[17] progress_1.2.3 curl_5.2.1
[19] broom_1.0.5 cellranger_1.1.0
[21] SparseArray_1.2.4 sass_0.4.9
[23] bslib_0.7.0 htmlwidgets_1.6.4
[25] plyr_1.8.9 zoo_1.8-12
[27] cachem_1.0.8 GenomicAlignments_1.38.2
[29] whisker_0.4.1 mime_0.12
[31] lifecycle_1.0.4 pkgconfig_2.0.3
[33] Matrix_1.6-5 R6_2.5.1
[35] fastmap_1.1.1 MatrixGenerics_1.14.0
[37] GenomeInfoDbData_1.2.11 shiny_1.8.1.1
[39] digest_0.6.35 colorspace_2.1-0
[41] ps_1.7.6 rprojroot_2.0.4
[43] crosstalk_1.2.1 RSQLite_2.3.6
[45] labeling_0.4.3 filelock_1.0.3
[47] fansi_1.0.6 timechange_0.3.0
[49] httr_1.4.7 abind_1.4-5
[51] compiler_4.3.2 bit64_4.0.5
[53] withr_3.0.0 backports_1.4.1
[55] BiocParallel_1.36.0 carData_3.0-5
[57] DBI_1.2.2 highr_0.10
[59] ggsignif_0.6.4 biomaRt_2.58.2
[61] MASS_7.3-60.0.1 DelayedArray_0.28.0
[63] rappdirs_0.3.3 rjson_0.2.21
[65] tools_4.3.2 interactiveDisplayBase_1.40.0
[67] httpuv_1.6.15 glue_1.7.0
[69] restfulr_0.0.15 callr_3.7.6
[71] promises_1.3.0 grid_4.3.2
[73] getPass_0.2-4 reshape2_1.4.4
[75] generics_0.1.3 gtable_0.3.4
[77] tzdb_0.4.0 data.table_1.15.4
[79] hms_1.1.3 xml2_1.3.6
[81] car_3.1-2 utf8_1.2.4
[83] XVector_0.42.0 BiocVersion_3.18.1
[85] pillar_1.9.0 later_1.3.2
[87] lattice_0.22-6 rtracklayer_1.62.0
[89] bit_4.0.5 tidyselect_1.2.1
[91] Biostrings_2.70.3 knitr_1.45
[93] git2r_0.33.0 ProtGenerics_1.34.0
[95] SummarizedExperiment_1.32.0 xfun_0.43
[97] matrixStats_1.3.0 DT_0.33
[99] stringi_1.8.3 lazyeval_0.2.2
[101] yaml_2.3.8 codetools_0.2-20
[103] evaluate_0.23 BiocManager_1.30.22
[105] cli_3.6.2 xtable_1.8-4
[107] munsell_0.5.1 processx_3.8.4
[109] jquerylib_0.1.4 Rcpp_1.0.12
[111] png_0.1-8 parallel_4.3.2
[113] XML_3.99-0.16.1 blob_1.2.4
[115] prettyunits_1.2.0 bitops_1.0-7
[117] viridisLite_0.4.2 crayon_1.5.2
[119] rlang_1.1.3 cowplot_1.1.3
[121] KEGGREST_1.42.0